
Software

POPSICLE
Population Size Coalescent-time-based Estimator
POPSICLE infers population size changes in the past by means of analyzing the distribution of coalescent times. The program is introduced in Gattepaille et al. (2016) and is available here as a .zip file. The folder contains the program and additional files such as example data. In addition, R should be installed on your system and the POPSICLE function can be found in popsicle.R.
CLUMPP
Cluster Matching and Permutation Program
CLUMPP is a program that deals with label switching and multimodality problems in population-genetic cluster analyses. CLUMPP permutes the clusters output by independent runs of clustering programs such as structure, TESS, or BAPS, so that they match up as closely as possible. The user has the option of choosing one of three algorithms for aligning replicates, with a tradeoff of speed and similarity to the optimal alignment. A program note describing CLUMPP was published in Jakobsson & Rosenberg (2007). CLUMPP was written by Mattias Jakobsson.
To be redirected to the CLUMPP page at the Rosenbergs Lab, Stanford University, click here.
MULTIORIGIN
MULTIple ORIGIN
MultiOrigin explores hypotheses about the number of origins and time of origin of species that are founded from an ancestral species through discrete events (i.e. polyploid species). The program uses a population divergence coalescence model together with a rejection sampling algorithm. The program was used in Jakobsson et al. (2006). MultiOrigin was written by Mattias Jakobsson.
To be redirected to the MultiOrigin page, click here.
COMPASS
COalescence siMulation PROGRAM ALLOWING SERIAL SAMPLES
The program COMPASS generates serial samples collected at various points in time. The samples are generated using coalescence simulations permitting various demographic scenarios. COMPASS uses an infinite sites model to generate polymorphism data for the samples. By generating serially sampled population-genetic data, COMPASS provide means of investigating the properties of data that has been collected at different time-points, and potentially, aid in interpreting the results from empirical data, for example polymorphism data collected from both present-day samples and historic samples. The program was written by Mattias Jakobsson and a program note was published in Jakobsson (2009).
To be redirected to the COMPASS page, click here.
MAXCON
MAXimum CONtribution from an ancestral population
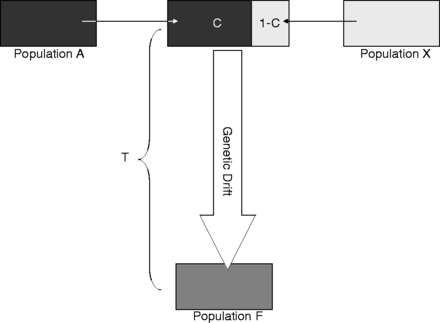
MAXCON is a program to estimate the maximum contribution from an ancestral population to a recent one. It uses bi-allelic markers sampled in two temporarily separated populations from which the time since divergence is known. In addition, some knowledge of the effective size of the contemporary population is required. MAXCON was described and tested in Sjödin et al. (2014) and a download is available here.
PMDTOOLS
POST Mortem Damage tools
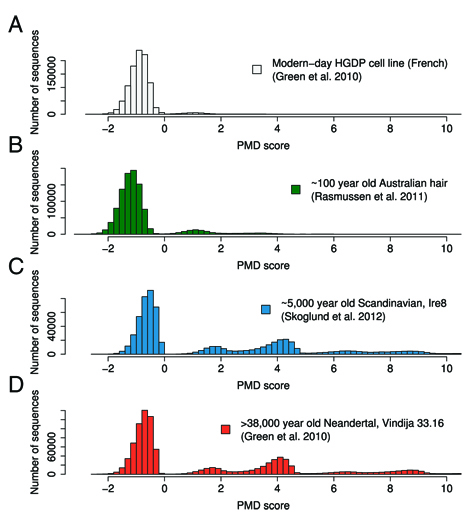
The program was developed to decipher endogenous, degraded DNA fragments from those derived from modern contaminations in ancient DNA extracts. It implements a likelihood framework utilizing patterns of postmortem damage, base quality scores and biological polymorphisms. The outcome is a sample specific PMD score that indicates, if it is genuinely of ancient material. The underlying methods were described in Skoglund et al. (2014) and the program itself can be downloaded, click here.
HAPLOPOP
HAPLOtypes for POPulation inference
The software helps to infer haplotypes within genomic windows of fixed size in order to improve accurate population assignment and structure inferences. HAPLOPOP utilizes the Gain of Informativeness for Assignment statistic that provides a criterion to decide whether markers should be combined into haplotypes. The software was introduced in Duforet-Frebourg et al. (2015), and is available here.
ADZE
Allelic Diversity analyZEr
ADZE is a program that implements the rarefaction method for analyzing allelic diversity across populations while correcting for sample size differences. Using individual multilocus genotype data on genetic polymorphisms, ADZE computes estimates of allelic richness, private allelic richness, and private allelic richness for combinations of populations. ADZE was written by Zach Szpiech. A description of the method was published in Szpiech et al. (2008). To be redirected to the ADZE page, click here.